
Цитокины адгезии при воспалении

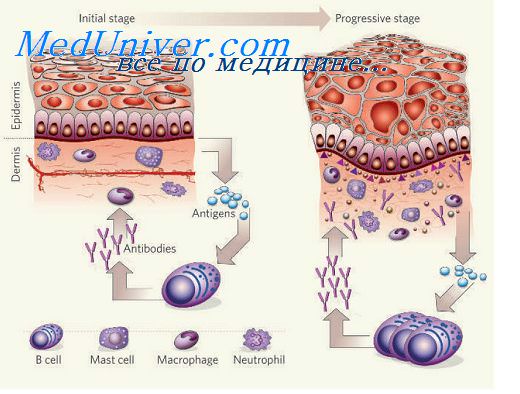
Молекулы адгезии. Роль молекул адгезии при воспалении.Движение лейкоцитов в очаг воспаления начинается с серии адгезионных событий, каждое из которых касается лейкоцитов определенного типа: нейтрофилов, моноцитов или лимфоцитов. Циркулирующие лейкоциты обычно вступают лишь в мимолетные контакты с эндотелиальными клетками посткапиллярных венул: лейкоциты как бы “скользят” по поверхности эндотелия сосудистой стенки. Эта фаза обеспечивается взаимодействием вначале Р-, а затем L- и Е-селектинов с углеводными компонентами мембран клеток. L-селектин экспрессирован на большинстве лейкоцитов. селектин эндотелиальных клеток опосредует адгезию нейтрофилов и моноцитов к эндотелию. Е-селектин экспрессируется на активированных эндотелиальных клетках и поддерживает адгезию лимфоцитов. Лигандами селектинов служат сиалил-фукозилированные олигосахариды в составе многих гликопротеинов и гликолипидов мембран клеток, например, муциноподобные молекулы. Муциноподобный домен содержит клеточная адгезионная молекула – мукозный адрессин (MAdCAM-1), которая за счет взаимодействия с L-селектином обеспечивает возврат лимфоцитов в мукозноассоциированную лимфоидную ткань. Фаза скольжения происходит без активации лейкоцитов, однако скользящие лейкоциты при контактах с поверхностью эндотелия получают сигналы активации, что ведет к их иммобилизации. Наступает вторая фаза прочной адгезии, опосредованная усилением способности лейкоцитарных интегринов связываться с лигандами из суперсемейства иммуноглобулинов на эндотелиальных клетках. В качестве сигналов активации могут служить воздействия цитокинов (хемокинов): MIP-ip, MCP-1, IL-8, MIF, PAF, С5а-фракции комплемента, которые способны связываться с глюкозамингликанами поверхности эндотелиальных клеток и действовать на “скользящие” лейкоциты.
Интегрины – это большое семейство молекул клеточной поверхности, представители которых обнаружены на большинстве типов клеток. Интегрины опосредуют взаимодействие клеток с их микроокружением, обеспечивая адгезию клетка – клетка и клетка – матрикс. Интегрины – это гетеродимеры гликопротеинов, состоящие из различных комбинаций а- и (J- цепей. Описано более 20 разных представителей интегринов. На лейкоцитах экспрессированы: LFA-1, Macl, pl50,95. Лигандами для LFA-1 являются : ICAM-1, ICAM-2, ICAM-3, для Macl -ICAM-1. Эти интегрины опосредуют адгезию к эндотелию нейтрофилов, базофилов, эозинофилов, моноцитов и лимфоцитов. В отличие от нейтрофилов остальные типы клеток могут адгезироваться к цитокин-активированным эндотелиальным клеткам через интегрины VLA-4 к лигандам VCAM-1. На поверхности эндотелиальных клеток лигандами интегринов служат молекулы, имеющие структурную гомологию с иммуноглобулинами. К ним относятся интерклеточные адгезионные молекулы: ICAM-1, ICAM-2, ICAM-3, васкулярно-клеточная адгезионная молекула – VCAM1. Последняя эксирессируется преимущественно на активированных эндотелиальных клетках. Следующая после прочной адгезии стадия трансмиграции лейкоцитов через эндотелий контролируется частично теми же интегринами, взаимодействующими с молекулами ICAM-1, расположенными и на внутренней, и на латеральной, и на базальной поверхности эндотелиальных клеток. Описаны и другие молекулы, облегчающие трансмиграцию лейкоцитов: например CD31 (РЕСАМ-1), обнаруженные и на эндотелиальных клетках, и на тромбоцитах, нейтрофилах, моноцитах, лимфоцитах. За трансмиграцию моноцитов отвечает интегрин CD18, но после активации эндотелиальных клеток под влиянием IL-1 и TNF-a трансмиграция идет при участии интегринов а, взаимодействующих с молекулой VCAM-1. Все стадии адгезии и трансмиграции зависят от активации эндотелиальных клеток, которая проявляется усилением экспрессии на них адгезионных молекул. Экспрессия Е-селектина усиливается в самые ранние стадии воспаления тромбином, гистамином или активированной системой комплемента, и не требует синтеза белка de novo. Роль стимуляторов на этой стадии могут играть различные оксиданты. – Также рекомендуем “Молекулы адгезии в подострой фазе воспаления.” Оглавление темы “Воспаление легких.”: |

Источник
Молекулы адгезии в подострой фазе воспаления.В подострой фазе воспаления активация эндотелиальных клеток опосредована, в основном, цитокинами: IL-1, TNF-a, которые индуцируют экспрессию Е-селектина, VCAM-1 и усиливают экспрессию ICAM-1. Такое же действие оказывает ЛПС. Эта активация связана с необходимостью синтеза белка de novo. Кинетика экспрессии трех вышеперечисленных молекул различна: TNF-a раньше всего усиливает экспрессию Е-селектина – за 2-4 часа до максимальной экспрессии VCAM-1 и ICAM-1. Только экспрессию ICAM-1 повышает IFNy, а IL-4 индуцирует только VCAM-1. Сочетанный эффект IFNy и IL-4 проявляется усилением и пролонгированием действия TNF-a в условиях хронического иммуно-опосредованного воспаления. Среди механизмов ингибиции тех же процессов активации эндотелиальных клеток описаны ингибирующие эффекты TGFfi, который ингибирует продукцию IL-8, снижает IL-8-зависимую трансмиграцию нейтрофилов через эндотелий, ингибирует экспрессию Е-селектина и адгезивность эндотелиальных клеток для нейтрофилов и лимфоцитов. Эффекты TGFp противоречивы: он стимулирует хемотаксис лейкоцитов, но препятствует их трансмиграции через эндотелий. С дефектом TGFp может быть связано развитие многофокусного воспаления с лейкоцитарной инфильтрацией многих тканей, с избыточной продукцией провоспалительных цитокинов. На эпителиальных клетках бронхов также экспрессированы адгезионные молекулы ICAM-1, экспрессия которых может повышаться под влиянием цитокинов IFNy, TNF-oc, IL-1(3. Экспрессия молекул ICAM-1 усиливается на эпителии при воспалении воздухоносных путей. На таких активированных эпителиальных клетках усилена адгезия эозинофилов, которая, в свою очередь, приводит к усиленной дегрануляции эозинофилов. Секретируемые при этом катионные белки могут участвовать в повреждении респираторного эпителия.
При остром воспалении легких нейтрофилы эмигрируют из альвеолярных капилляров и двигаются в ответ на воспалительные стимулы, преодолевая интерстициальный матрикс между эндотелием капилляров и альвеолярным эпителием. Этот процесс включает адгезию нейтрофилов и их локомоции. Альвеолярный интерстиций состоит из фибробластов. Адгезия нейтрофилов к альвеолярным фибробластам повышается под влиянием PAF за счет повышения экспрессии интегринов (CD11/CD18). Миграционная активность нейтрофилов возрастала под влиянием хемоаттрактанта IL-8, который секретировали альвеолярные фибробласты, активированные под влиянием TNF-oc. Фибробласты служат не только субстратом адгезии, но и источником стимулирующих молекул, обеспечивая миграцию нейтрофилов через воспаленный альвеолярный интерстиций. Для бактериальной пневмонии характерен ранний выход нейтрофилов в интерстиций и в просвет альвеол. При этом контакт нейтрофилов с фибробластами опосредован взаимодействием интегринов (CD11/ CD18) с молекулами ICAM-1(CD54), экспрессированными на фибробластах. Конститутивная экспрессия адгезионных молекул ICAM-1 на альвеолярных эпителиальных клетках в 22 раза выше, чем на эндотелиальных кчетках капилляров. ICAM-1 экспрессированы, в основном, на пневмоцитах типа I, а не на их предшественниках пневмоцитах типа II. При пневмонии экспрессия ICAM-1 повышается в 178 раз на пневмоцитах типа II, т.е. происходит их ускоренная дифференцировка в ответ на повреждение эпителия альвеол. Кроме того, непосредственно в очаге пневмонии были обнаружены нейтрофилы с измененной экспрессией адгезионных молекул: L-селектина и CD18. Эмиграция нейтрофилов в просвет альвеол при пневмонии, вызванной Streptococcus pneumoniae, связана со снижением экспрессии L-селектина и повышением экспрессии CD18. Нейтрофилы в воздушном пространстве альвеол легкого имеют повышенный уровень экспрессии адгезионных молекул CD18, что может способствовать фагоцитозу патогенных бактерий. Адгезия моноцитов к эндотелиальным клеткам легочных капилляров также опосредована взаимодействием интегринов на моноцитах с молекулами ICAM на эндотелиальных клетках. Аккумуляции моноцитов в легких способствует и то, что диаметр легочных капилляров (6-8um) меньше диаметра многих моноцитов крови (7-10um), поэтому прохождение моноцитов через легочные капилляры требует деформации, пластичности моноцитов, которая в первую очередь нарушается при воспалении. Моноциты застревают в узких легочных капиллярах, прилипают к эндотелиальным клеткам и проделывают трансмиграцию в просвет альвеол. Бактериальные компоненты и продукты (в частности ЛПС) повышают адгезивность моноцитов, усиливая экспрессию адгезионных молекул. Адгезионные молекулы могут стать клинически значимыми мишенями для противовоспалительной терапии. В настоящее время ведется поиск агентов, селективно ингибирующих адгезионные молекулы. – Вернуться в оглавление раздела “Пульмонология.” Оглавление темы “Воспаление легких.”: |
Источник
Роль цитокинов при воспалении в легких.Начальный местный эффект провоспалительных цитокинов – это инициация воспаления за счет расширения сосудов, усиления местного кровотока (жар, краснота) и повышения их проницаемости, которые ведут к накоплению экссудата (отек, боль). Последующий местный эффект провоспалительных цитокинов – это индукция экспрессии на эндотелиальных клетках адгезионных молекул, связывающих циркулирующие в крови лейкоциты и способствующих их миграции из капилляров в ткани. Взаимодействие специальных адгезионных молекул на лейкоцитах и на эндотелиальных клетках позволяет лейкоцитам преодолевать барьер сосудистой стенки путем диапедеза. Дальнейшая миграция лейкоцитов через ткани в очаг инфекции или воспаления контролируется специальными молекулами – белками низкой молекулярной массы – хемокинами, которые продуцируются и секретируются не только активированными макрофагами, но и эндотелиальными клетками, фибробластами, гладкомышечными клетками. Их основная функция – служить для лейкоцитов хемоаттрактантами, рекрутировать их в очаг воспаления. По ходу миграции лейкоцитов хемокины оказывают на них и активирующее действие. Наряду с местными эффектами провоспалительные цитокины вызывают ряд системных реакций. В качестве эндогенных пирогенов, ответственных за развитие лихорадки, выступают IL-1 и IL-6. Те же цитокины индуцируют синтез белков острой фазы: С-реактивного белка (CRP), сывороточного амилоида Р, манноза-связывающего белка. Одним из системных эффектов провоспалительных цитокинов является индукция лейкоцитоза, т.е. увеличение количества циркулирующих лейкоцитов за счет ускорения их выхода из костного мозга и депо, где они могут длительно сохраняться в состоянии пристеночной адгезии.
Параллельно провоспалительные цитокины индуцируют на эндотелиальных клетках экспрессию молекул, запускающих процессы свертывания крови, что способствует формированию тромба, препятствующего распространению инфекции через кровеносное русло. Провоспалительные цитокины рекрутируют в очаг воспаления не только фагоцитирующие клетки, но и лимфоциты, в частности Т-лимфоциты, которые под влиянием тех же цитокинов активируются и начинают сами продуцировать и секретировать цитокины: IL-2, IL-4, IL-10, IL-13, IFNy и другие. Среди перечисленных цитокинов есть как провоспалительные (IFNy), так и противовоспалительные: IL-4, IL-10. Противовоспалительные цитокины могут ингибировать продукцию и секрецию провоспалительных цитокинов на более поздних этапах воспаления, когда включаются механизмы негативной регуляции с обратной связью. На модели интерстициального воспаления легких было показано, что вначале развивается диффузный альвеолит, который характеризуется рекрутированием в альвеолы и активацией макрофагов, гранулоцитов и лимфоцитов. Вслед за этим ранним воспалительным ответом усиливается пролиферация фибробластов и синтез компонентов внеклеточного матрикса. Во всех этих процессах участвуют цитокины: TNF-a, IL-1, MIP-1. Воспаление легкого заканчивается фиброзом за счет резкого увеличения содержания в легочной ткани гидроксипролина, стимуляции пролиферации фибробластов и депозиции белков внеклеточного матрикса. В динамике процесса воспаления легких аккумуляция и активация лейкоцитов ведут в конечном итоге к фиброзу. В участках активного фиброзирования легочной ткани выявляются цитокины, продуцируемые, в частности, легочными эозинофилами: IL-1, TGFp, IL-3, IL-5, IL-6, IL-8. Поддержание иммунного гомеостаза легких зависит от баланса достаточно эффективных защитных механизмов, обеспечивающих элиминацию вдыхаемых патогенов, и не менее эффективных механизмов ингибиции воспаления для ограничения последующего повреждения ткани легкого. – Также рекомендуем “Роль лимфоцитов при воспалении в легких.” Оглавление темы “Патогенез воспаления в легких.”: |

Источник
Цитокины при воспалении. Миграцию ингибирующий фактор.Миграцию ингибирующий фактор (MIF) был впервые описан в 60-е годы как продукт активированных Т-лимфоцитов. Только через 25 лет удалось клонировать соответствующий ген, получить рекомбинантный белок и специфические моноклональные антитела. Биологическая активность MIF может быть охарактеризована как негативный хемотаксический эффект: торможение миграции фагоцитирующих клеток (гранулоцитов, моноцитов, макрофагов). Благодаря такому действию этот цитокин участвует в мобилизации фагоцитирующих клеток в очаг инфекции или воспаления на последнем этапе аккумуляции клеток в очаге. Кроме того, у MIF описаны и другие свойства провоспалительного цитокина. Наряду с TNF-a и IL-1 он участвует в каскаде реакций эндотоксического шока, возможно, контролируя уровень TNF-a. Этот цитокин участвует в качестве эффекторной молекулы в развитии клеточного иммунного ответа, реакций ГЗТ. Уровень продукции MIF, как правило, повышается при инфекциях и воспалительных процессах.
Изучение способности мононуклеаров крови к усиленной продукции MIF давно используется в качестве одного из тестов для оценки функциональной активности Т-лимфоцитов и специфической сенсибилизации клеток (реакция торможения миграции лейкоцитов -РТМЛ). В последние годы показано, что продуцентами MIF кроме активированных Т-лимфоцитов могут быть моноциты и макрофаги, которые отвечают продукцией и секрецией MIF, наряду с другими провоспалительными цитокинами, на индукцию ЛПС. Кроме того, пресинтезированный MIF был обнаружен в передней доле гипофиза и была показана способность клеток передней доли гипофиза отвечать продукцией MIF на индукцию ЛПС. Усиленную секрецию цитокина in vivo вызывал кортикотропин-релизинг фактор (CRF), что было расценено как компонент стрессорной реакции. В связи с. этим возникло предположение о том, что MIF может выполнять функции контр-регулятора иммунного ответа по отношению к глюкокортикоидам, которые известны как наиболее сильные ингибиторы воспаления и клеточного иммунного ответа. В физиологических концентрациях глюкокортикоиды индуцируют секрецию MIF макрофагами и Т-лимфоцитами, хотя секрецию других провоспалительных цитокинов те же глюкокортикоиды подавляют. Очевидно, MIF контролирует противовоспалительные эффекты глюкокортикоидов. Так, например, MIF блокировал протективный эффект дексаметазона на модели эндотоксического шока. Показана способность MIF противостоять ингибирующему действию глюкокортикоидов на секрецию макрофагами провоспалительных цитокинов: TNF-а, IL-1, IL-6, IL-8. Уровень MIF может повышаться как следствие глюкокортикоидной терапии. Повышенный уровень MIF контролирует иммуносупрессирующие эффекты глюкокортикоидов: эндогенных или введенных для лечения. Отсюда анти – MIF стратегия может быть полезна для повышения иммуносупрессивного и противовоспалительного действия глюкокортикоидов. – Также рекомендуем “Интерферон – гамма. Значение и функции интерферона гамма при воспалении.” Оглавление темы “Воспаление легких.”: |
Источник