
Фактор хагемана в воспалении

Фактор Хагемана (XII фактор свертывания крови) представляет собой белок, который участвует в двух важнейших биологических процессах – гемокоагуляции и образовании кининов. Кинины можно рассматривать как относительно низкомолекулярные активные медиаторы воспаления, обладающие одновременно и другими биологическими свойствами (например, способностью вызывать бронхоспазм). Фактор Хагемана оказывается связующим звеном между процессами воспаления и свертывания крови; до его открытия связь этих процессов была гипотетической.
В крови фактор Хагемана циркулирует в неактивной форме. Он активируется при контакте с отрицательно заряженными поверхностями, к которым, в частности, относятся стекло, эластин, почечная основная мембрана и, что особенно важно для ревматологии, суставной хрящ, урат натрия, пирофосфат кальция.
Активированный фактор превращает циркулирующий в крови малоактивный прекалликреин в активный фермент калликреин, который взаимодействует с находящимися в плазме неактивными предшественниками кининов – низкомолекулярным и высокомолекулярным кининогенами. Из последних в результате такого химического взаимодействия образуются кинины, наиболее известным представителем которых является брадикинин.
Кроме того, активированный фактор Хагемана взаимодействует с добавочным количеством его неактивированных молекул, и за счет этого происходит активирование плазменного предшественника тромбопластина (ППТ), который переводит определенное количество протромбина в тромбин и в конечном итоге превращает фибриноген в фибрин, т. е. таким образом осуществляется процесс свертывания крови. Система фактора Хагемана эффективнее активирует калликреин, чем ППТ.
Существуют многообразные пути взаимодействия как между системами кининобразования и свертывания крови, так и внутри каждой из этих систем. Так, калликреин не только участвует в генерации кининов, но и способствует превращению плазминогена в протеолитический фермент плазмин (фибринолизин), расщепляющий, в частности, молекулы недавно образовавшегося фибрина. Считается, что в организме плазмин не образуется без активации фактора Хагемана.
Последний, таким образом, участвует не только в процессе свертывания крови, но и в продукции одного из активных противосвертывающих факторов. Плазмин в свою очередь способен активировать фактор Хагемана. Таким же свойством обладают калликреин (причем эта реакция ускоряется высокомолекулярным кининогеном) и комплекс прекалликреина с высокомолекулярным кининогеном. Примечательно, что активированный плазменный предшественник тромбопластина (ППТа), усиливая процессы свертывания, в то же время превращает плазминоген в плазмин, т. е. уменьшает коагуляционные свойства крови. Этим, по-видимому, объясняются большие возможности для саморегулирования процесса гемокоагуляции.
Система фактора Хагемана генерирует ряд продуктов, имеющих непосредственное отношение к развитию воспалительного процесса. Основным кинином считается брадикинин, который повышает проницаемость капилляров, расширяет артериолы, способствует освобождению гистамина из тучных клеток и повышает синтез простагландинов, рассматриваемых в настоящее время как важные медиаторы воспаления. Калликреин и фибринопептиды, возникающие в результате ферментативного расщепления фибриногена плазмином, усиливают хемотаксис лейкоцитов. Плазмин, кроме того, активирует первый компонент комплемента, расщепляет его третий компонент, превращает проколлагеназу синовиальных клеток в коллагеназу, обладающую деструктивным влиянием на ткани.
Известны физиологические антагонисты ряда биологически активных веществ, участвующих в рассматриваемой системе. Комплекс антитромбина III с гепарином угнетает фактор Хагемана и ППТа. Циркулирующий в крови a2-макроглобулин ингибирует плазмин и калликреин, а a1-антитрипсин-плазмин и ППТа. Активность брадикинина тормозится ферментом кининазой I (она угнетает также активность таких компонентов комплемента, как СЗа и С5а).
Изложенные факты демонстрируют предпосылки для возможных сочетаний воспалительного процесса с расстройствами гемокоагуляции. Последние в виде тромбирования микроциркуляторного русла часто наблюдаются в очагах воспаления. В норме деятельность систем кининообразования и гемокоагуляции протекает сбалансированно, но при качественных или даже чисто количественных изменениях их компонентов равновесие нарушается.
Опубликовал Константин Моканов
Источник
Здравствуйте! Сегодня мы поговорим о 12-ом факторе свертываемости крови, или о факторе Хагемана.
История
В 1950-е годы анализ крови перед операцией выявил удлинение внутреннего пути свертывания у железнодорожника Джона Хагемана. Знаменитый ученый Оскар Ратноф (Oscar Ratnoff) начал исследовать эту кровь и обнаружил у Хагемана дефицит неизвестного фактора свертываемости, который был назван фактором Хагемана, а потом – фактором XII.
У железнодорожника было резкое удлинение АЧТВ, и при этом не было никаких признаков кровоточивости. Интересно,что при этом умер Джон Хагеман не от кровотечения, а от тромбоза – в результате тромбоэмболии легочной артерии в молодом возрасте.
Фактор XII
Фактор XII – это профермент, синтезируемый печенью и циркулирующий в крови. Активация FXII происходит при контакте с различными поверхностями, даже со стеклом, в организме чаще с поврежденными поверхностями.
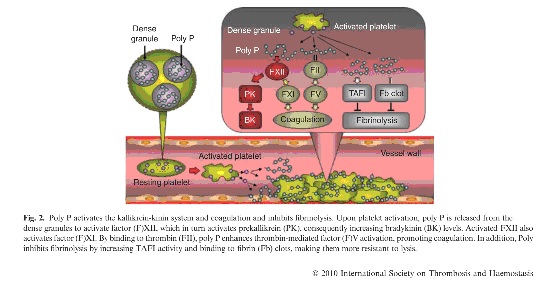
Это очень хорошая картинка, взятая из статьи, посвященной Оскару Ратнофу, обнаружившему неизвестный доселе фактор свертывания крови у сцепщика Джона Хагемана. Она иллюстрирует запуск “контактного” (“внутреннего”) пути.
-Активация тромбоцита приводит к выбросу из него гранул с полифосфатами.
-Полифосфаты запускают активацию фактора Хагемана на поверхности образующегося агрегата тромбоцитов, с последующим запуском всей цепочки.
-Потом полифосфаты вымываются и фактор Хагемана становится одним из основных факторов фибринолиза.
Дефицит уровня FXII
Дефицит уровня FXII – самая частая причина удлинения АЧТВ. И если причиной является такой дефицит, добавление в систему нормальной плазмы, содержащей нормальные количества фактора XII, укорачивает и приближает к норме АЧТВ.
Напомним, что в АЧТВ для активации свертывания используется “частичный тромбопластин”, то есть это тромбопластин без тканевого фактора, фактически мы говорим о фосфолипидах.
Фосфолипиды в системе активации АЧТВ играют роль поверхности тромбоцитов. Если в системе есть антитела против фосфолипидов, то, соответственно, и в этом случае АЧТВ будет замедляться – это и есть то, что получило название Волчаночного Антикоагулянта (ВА).
Таким образом, ВА является второй причиной удлинения АЧТВ. В присутствии ВА АЧТВ плохо корригируется добавлением плазмы, богатой фактором XII, потому что его и так много в системе.
А если мы бездумно блокируем тромбин гепарином, то будут удлиняться показатели как внешнего, так и внутреннего пути. Поэтому на фоне активной терапии гепаринами АЧТВ тоже будет удлиняться, в данном случае, фармакологически.
Главное и ключевое то, что при дефиците FXII повышается риск не кровотечений, а тромбозов! Почему? Потому что фактор XII в гораздо большей степени активирует фибринолиз, чем тромбоз.
Один из главных рисков венозного тромбоза.- недостаточный фибринолиз. Поэтому, Джон Хаггеман умер не от кровотечения, а от тромбоза.
По своей химической структуре FXII очень похож и эволюционно связан с tPA (тканевым активатором плазминогена). При этом следует помнить, что концентрация FXII значительно превышает концентрацию tPA в крови.
Для коагуляции снижение концентрации FXII не критично, а вот для фибринолиза — может быть катастрофично.
Мы видим парадокс: удлинение АЧТВ очень часто указывает на дефицит фактора Хагемана, и в пробирке мы видим гипокоагуляцию. А в организме беременной женщины мы видим гипофибринолиз – резкое торможение фибринолиза. Фибриноид начинает накапливаться, плацента быстро стареет, тромбируются мелкие вены, резко ухудшается плацентарное кровообращение с материнской стороны, и плод страдает.
Диагностика дефицита фактора 12
Определяемый нами полиморфизм rs1801020 (C46T) ассоциирован с резким снижением продукции печенью FXII (сам фактор совершенно нормальный).
В мировой научной среде проводилось очень много исследований по поводу дефицита фактора XII на течение беременности. Немцы из Геттингентского университета установили жесткую связь между наличием этого полиморфизма у женщин с привычным невынашиванием беременности (3 остановки подряд и более). А японские коллеги из Токийского университета, выявили связь этого полиморфизма с многократными неудачными попытками ЭКО.
Сейчас абсолютно доказанными вещами является то, что дефицит фактора 12 – гипофибринолиз – связан с нарушением плацентарной функции, невынашиванием на ранних сроках, ранней преэклампсией, неоднократными неудачными попытками ЭКО
Самое резкое снижение фактора XII мы наблюдаем у гомозигот T/T. У гетерозигот (около 34 %) имеется промежуточное значение концентрации FXII.
“Хороший вариант” гомозигот встречается примерно в 60%.
Посмотрите, пожалуйста, вот очень важная схема:
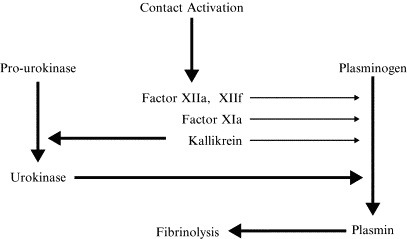
Что мы наблюдаем?
-Фактор XII активирует калликреин, который мощно активирует урокиназу.
-Урокиназа запускает образование плазмина из плазминогена. Это главный путь стимуляции фибринолиза фактором XII.
-Параллельно работают другие механизмы, не главные, но активно вносящие свой вклад в активацию плазмина.
-Сам активированный FXII катализирует переход плазминогена в плазмин.
-Активированный фактор XII активирует фактор XI (основной механизм запуска “внутреннего пути свертывания”). Но активированный таким образом FXI не только запускает генерацию тромбина, но и генерацию плазмина.
-И, наконец, активированный калликреин не только активирует урокиназу, но и сам, напрямую – плазмин.
Фактор 12 при АФС
При АФС (антифосфолипидный синдром) могут быть и АТ, направленные против FXII. Это важный анализ, но к сожалению, в нашей стране он не проводится (реактивы не серцифицированы).
При наличии антифосфолипидных АТ как правило наблюдается резкое снижение FXII , поэтому ,всегда внимательно оцениваем АЧТВ, которое часто начинает удлиняться именно при снижении концентрации FXII.
FXII может быть мишенью аутоиммунной атаки при АФС. Это частично объясняет механизм действия волчаночного антикоагулянта, удлиняющего АЧТВ.
Всё это указывает на то, что нарушение фибринолиза, одним из важных маркеров которого является снижение уровня FXII и связанное с этим удлинение АЧТВ, играет очень важную роль в нарушении плацентарной функции при АФС.
Дефицит 12 фактора у беременных
Дефицит фактора ХII может приводить к очень серьезным проблемам во время беременности, начиная от невынашиваемости на ранних сроках, и заканчивая задержкой развития плода и антенатальной гибелью.
Часто большое значение имеют не изолированные факторы, а связки полиморфизмов. В частности, при связке с таким полиморфизмом, как PAI-1, особенно если речь идет о гомозиготе 4G/4G, мы будем наблюдать достаточно серьезные проблемы с плацентацией. Потому что очистка от фибрина зоны инвазии экстравиллезного трофобласта с стенках спиральных артерий имеет очень большое значение, практически решающее для того, чтобы плацентация прошла хорошо.
Ассоциации с нарушениями фибринолиза
1) Если говорить об опасных связках, это такие полиморфизмы, как PAI-1, FXI, FXIII, FXII, PLAT и ген фибриногена. Если говорить о полиморфизмах сосудистого тонуса, это ангиотензинпревращающий фермент.
2) Группы крови (все плохие, кроме первой); Частота аллелей для европейской популяции:
3) Если говорить об антителах к фосфолипидам, то АТ против бета2GP1 и аннексина V, двух факторов, которые играют важную роль в нормальном фибринолизе.
4) Нерастворимые комплексы фибрин-мономеров (РКФМ). То есть это уже фибрин, только не успевший дальше полимеризироваться и пока еще растворимый. Этот показатель напрямую связан с активностью тромбина, и значительное повышение – плохо. Если первая цифра 4 (4,0-4,9) — нормально для беременности, они всегда более высокие с конца первых недель беременности, но если больше 5 — значит высокая активность тромбина, значит его лучше аккуратно притушить назначением гепаринов.
Для того, чтобы не пропустить серьёзные осложнения беременности, необходимо комплексное обследование комбинации гемостаза , диагностики аутоиммунных проблем иполного спектра полиморфизмов гемостаза.
Источник
Добавил:
Upload Опубликованный материал нарушает ваши авторские права? Сообщите нам.
Вуз:
Предмет:
Файл:
Патофизиология 1 коллоквиум.docx
Скачиваний:
186
Добавлен:
22.03.2016
Размер:
264.96 Кб
Скачать
Эффекты фактора Хагемана
Плазминоген Фибринолиз дериваты фибрина | Плазмин активация комплемента анафилактоксин (С4а, С3а, С5а) | Калликреиноген калликреин 2-глобулин (кининоген) брадикинин | Свертывание крови образование сгустков крови |
Роль системы комплемента в развитии воспаления
Анафилатоксины (С4а, С3а, С5а) | С3b | C5b-9 | ||
Активация тканей синтез ПГ и ЛT спазм гладкой мускулатуры бронхоспазм | Дегрануляция лаброцитов высвобождение гистамина повышение проницаемости стенки сосудов отек | Хемотаксис лейкоцитов Активация иммунного ответа Угнетение иммунного ответа | Опсонизация Активация фагоцитоза | Образование каналов в клеточных мембранах |
Механизмы возникновения и действия плазменных и клеточных медиаторов
Плазма крови | Активация XII (ф Хагемана) | Система свертывания крови и фибринолиза | Продукты распада фибрина | |||
Калликреин -кининовая система | Брадикинин | |||||
Активация системы комплемента | С3а, С5а | |||||
проницаемости стенки сосуда | отёк | |||||
Клетки | – Дегрануляция тучных клеток и базофилов | Гистамин | ||||
– Тромбоциты | Серотонин | |||||
– Фагоциты | Лизосомальные ферменты катионные белки Супероксидные радикалы Окcид азота | |||||
– Поврежденные клетки тканей | Фактор активации тромбоцитов Простагландины Лейкотриены | |||||
12.2.1.3. Сосудистые изменения при воспалении:
кратковременное сужение сосудов – ишемия | тонуса вазоконстрикторов, действие норадреналина |
артериальная гиперемия | тонуса вазодилататоров; паралич вазоконстрикторов; действие медиаторов (гистамин, лейкотриены), К+, Н+ на сосудистую стенку |
венозная гиперемия | внутрисосудистые факторы: сгущение крови; образование микротромбов; пристеночное стояние лейкоцитов; набухание форменных элементов крови и стенки сосудов в кислой среде. внесосудистые факторы: сдавление стенок венозных и лимфатических сосудов экссудатом и клеточным инфильтратом; разрушение соединительнотканных волокон, окружающих стенки капилляров и венул. |
стаз | застойный, истинно-капиллярный |
12.2.2. ЭКССУДАЦИЯ – выход белоксодержащей жидкой части крови и форменных элементов в очаг воспаления
ПАТОГЕНЕЗ ЭКССУДАЦИИ:
повышение проницаемости сосудов.
| гидростатического давления в капиллярах и венулах
| осмотического и онкотического давления в очаге воспаления – электролитов и белка в тканях |
Виды экссудатов:
Серозный | Фибринозный | Геморрагический |
(ожоговое, вирусное, аллергическое воспаление) Содержит 2-3% белка (альбумины), небольшое количество лейкоцитов, прозрачен | Образуется при значительном повреждении эндотелия. Содержит фибриноген, который при соприкосновении с тканями превращается в фибрин (при дифтерии, дизентерии) | Образуется при тяжелых повреждениях сосудов с разрушением базальной мембраны. Характерен для гриппозной пневмонии, сибирской язвы. Содержит значительное число эритроцитов |
Гнойный | Гнилостный | Смешанный |
Разделяется при центрифугировании на гнойную сыворотку и осадок. В осадке – гнойные тельца, разрушенные ткани, микробы. В сыворотке – гидролитические ферменты лизосом лейкоцитов. Способен лизировать окружающие ткани | При попадании анаэробной инфекции |
Значение экссудации:
Положительное: | Отрицательное: |
– локализация процесса |
|
Эмиграция лейкоцитов – выход лейкоцитов в очаг воспаления
Стадии эмиграции:
1. Краевое стояние лейкоцитов у внутренней стенки сосудов и роллинг (качение) | 2. Выход лейкоцитов через стенку сосуда | 3. Движение лейкоцитов в очаге воспаления |
| Патогенез:
|
|
Соседние файлы в предмете Патологическая физиология
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
Представлен клинический случай редкого заболевания – болезни Хагемана – нарушение гемостаза, характеризующееся значительным снижением активности плазменного фактора XII свертывания крови,у пациентки 58 лет с кардиальной патологией.
Болезнь Хагемана (или дефект Хагемана, или дефицит фактора XII свертывания крови) – крайне редкое нарушение гемостаза, характеризующееся значительным снижением активности плазменного фактора XII
(ФXII, фактор Хагемана) свертывания крови [1]. Частота встречаемости болезни Хагемана в популяции не превышает 1 случая на 1 млн населения [2]. В большинстве случаев данный дефект наследуется по рецессивно-аутосомному типу, однако в некоторых семьях выявлено аутосомно-доминантное наследование [1, 2]. По-видимому, синтез фактора XII свертывания крови контролируется двумя аутосомными генами (бимодальный тип наследования). Ген, кодирующий синтез фактора XII свертывания крови, локализуется в 5-й хромосоме [1].
ФXII – сиалогликопротеин (молекулярная масса
около 82 кДа), плазменный профермент, место синтеза которого в организме точно не установлено [3]. ФXII подвержен контактной и ферментной активации, в т. ч. адреналином, калликреином в присутствии отрицательно заряженного высокомолекулярного кининогена, фактором XI, а следовательно, играет ключевую роль в функционировании не только свертывающей, но и калликреин-кининовой, а также фибринолитической систем гемостаза [4].
Система гемостаза филогенетически предназначена для остановки кровотечения при травме и обеспечения жидкого состояния крови при сохранении биологической целостности сосудистой стенки [5]. С современных позиций различают первичный, или сосудисто-тромбоцитарный, и вторичный, или коагуляционный, гемостаз. Первичный гемостаз обусловлен спазмом сосудов и их механической закупоркой агрегатами тромбоцитов, вторичный представляет собой сложный ферментный процесс, в котором участвуют ряд протеолитических ферментов, а также неферментные белковые и фосфолипидные компоненты [1, 5]. Коагуляционный гемостаз может протекать по внешнему и внутреннему механизму, активатором последнего и служит ФXII [2, 4]. Завершающим этапом работы системы гемостаза является фибринолиз – расщепление фибрина под воздействием плазмина, в результате чего происходят разрушение фибринового сгустка и реканализация сосудов после остановки кровотечения [1, 5]. Между процессами свертывания крови и фибринолиза в организме постоянно поддерживается равновесие. Фибринолиз также может протекать по внешнему или внутреннему механизму [2, 4]. Внешний путь активации осуществляется при участии тканевых активаторов, таких как тканевый активатор плазминогена и урокиназа, синтезируемых непосредственно в эндотелии сосудов. Внутренний механизм активации осуществляется благодаря плазменным активаторам (Хагеман-зависимый путь) и активаторам форменных элементов крови (Хагеман-независимый путь). Хагеман-зависимый фибринолиз происходит под влиянием ФXII свертывания крови, который и вызывает превращение неактивного плазминогена в плазмин [1, 2, 5].
Экспериментальные исследования и клинические наблюдения показали, что роль ФXII более значима именно для процессов фибринолиза, нежели для коагуляционного ответа, поэтому при наличии у человека дефекта Хагемана отсутствуют какие-либо геморрагические проявления и отмечается высокая предрасположенность к тромбозам [2, 4, 6].
Впервые дефицит коагуляционного ФXII был выявлен в 1955 г. O.D. Ratnoff и J.E. Colopy, которые представили клиническое наблюдение 37-летнего пациента Джона Хагемана, по имени которого и были названы отсутствующий у него плазменный фактор свертывания и дефект системы гемостаза, обусловленный врожденной недостаточностью этого протеина [7]. Пациент умер от развившейся на фоне длительной иммобилизации тромбоэмболии легочной артерии после перелома тазовых костей.
В настоящее время в мире имеется описание чуть более 100 случаев этого заболевания [8]. Представляем собственное клиническое наблюдение пациентки кардиологического отделения с выявленным дефектом Хагемана.
Пациентка М., 58 лет,
поступила в отделение нарушения ритма клинического кардиологического диспансера (ККД) г. Омска с жалобами на одышку смешанного характера при подъеме до 3-го этажа, повышение артериального давления (АД) до 180/100 мм рт. ст., постоянные перебои в работе сердца, усиливающиеся при эмоциональных и физических нагрузках, отеки стоп и голеней к вечеру.
Из анамнеза заболевания известно, что считает себя больной в течение 5 лет с 2011 г., когда появились перебои в работе сердца, одышка при переносимой ранее физической нагрузке. При записи электрокардиограммы (ЭКГ) нарушения ритма не выявлялись, наблюдалась у терапевта по месту жительства с диагнозом соматоформной дисфункции вегетативной нервной системы.
С конца 2012 г. отметила ухудшение состояния, участились перебои в работе сердца, значительно снизилась толерантность к физическим нагрузкам, появилось ощущение постоянного дискомфорта за грудиной. Для уточнения диагноза и подбора терапии пациентка направлена в отделение нарушения ритма ККД.
При проведении дообследования в условиях стационара были получены следующие результаты: в общем анализе крови отмечалось увеличение скорости оседания эритроцитов (29 мм/ч); в биохимическом анализе сыворотки крови – повышение уровней общего билирубина (22,7 мкмоль/л), преимущественно за счет непрямой фракции (17,7 мкмоль/л), аланиновой аминотрансферазы (87 Е/л), общего холестерина (5,8 ммоль/л) и триглицеридов (4,52 ммоль/л). ЭКГ: ритм синусовый, с частотой сердечных сокращений (ЧСС) 52 в минуту, электрическая ось сердца не отклонена, признаки дилатации левого предсердия, синдром ранней реполяризации желудочков. Холтер-мониторирование ЭКГ: основной ритм синусовый с ЧСС 52-74-128 в минуту, прирост ЧСС на нагрузку достаточный, снижение ЧСС в ночные часы достаточное, вариабельность сердечного ритма сохранена, зарегистрированы 17 предсердных экстрасистол, 1 парная, 1 неустойчивый пароксизм наджелудочковой тахикардии, 7 эпизодов диагностически значимой депрессии сегмента ST по каналам 2 и 3 (общей продолжительностью 42 мин 11 с), без отметки в дневнике о болевых ощущениях. Эхокардиография: корень аорты – 3,2 см, восходящий отдел аорты – 3,0 см, дуга аорты – 2,4 см; левое предсердие – 3,6×4,6 см; конечный диастолический размер левого желудочка – 4,8 см; конечный систолический размер левого желудочка – 3,1 см; конечный диастолический объем – 109 мл; конечный систолический объем – 38 мл; толщина межжелудочковой перегородки – 0,9 см; толщина задней стенки левого желудочка – 0,9 см; правый желудочек – 2,5 см; правое предсердие – 3,2×3,8 см; фракция выброса – 65%; аортальный клапан – уплотнены фиброзное кольцо и створки, раскрытие створок – 2,0 см; митральный клапан – м-образный, движение створок разнонаправленное, створки уплотнены; регургитация через митральный клапан – 1 степени, регургитация через трикуспидальный клапан – 1 степени. Чреспищеводная электростимуляция сердца: ишемические изменения. Ультразвуковое исследование органов брюшной полости: признаки жирового гепатоза, диффузных изменений поджелудочной железы.
Был установлен диагноз: ишемическая болезнь сердца. Стенокардия напряжения, функциональный класс III. Артериальная гипертензия, стадия III, риск 4. Пароксизмальная наджелудочковая тахикардия. Синдром ранней реполяризации желудочков. Хроническая сердечная недостаточность II А, функциональный класс II. Неалкогольная жировая болезнь печени: стеатогепатит, активность 1.
На фоне проведенного лечения (диета № 10; соталол 0,08 г по 1½ таблетки утром и вечером под контролем АД, ЧСС, ЭКГ; лизиноприл 0,005 г по ½ таблетки вечером; урсодезоксихолевая кислота 0,25 г по 2 капсулы на ночь) самочувствие пациентки улучшилось – уменьшились частота и интенсивность боли за грудиной, уровень АД снизился до 135/80 мм рт. ст. Для уточнения данных о состоянии желудочков сердца, в т. ч. их сократительной способности, было рекомендовано проведение коронарной вентрикулографии (КВГ).
Перед подготовкой к плановому диагностическому эндоваскулярному вмешательству у больной было выявлено удлинение АЧТВ до 100 с неясной этиологии. После консультации гематолога, который заподозрил наличие коагулопатии – гипокоагуляции во внутреннем механизме свертывания (дефицит факторов свертывания XI, XII?), возможно, вторичного характера на фоне патологии печени, пациентка была направлена на обследование в ФБГУ «Гематологический научный центр» (ГНЦ) Министерства здравоохранения Российской Федерации (Москва).
При сборе анамнеза в ГНЦ установлено, что ранее повышенной кровоточивости у пациентки не наблюдалось. Наследственный анамнез по заболеваниям системы гемостаза не отягощен. В коагулограмме было выявлено: время свертывания – 10 мин 13 с, АЧТВ – более 250 с (норма – до 38 с), протромбиновый индекс – 80% (норма – 90-105%), фактор VIII – 70% (норма – 50-200%), фактор IX – 70% (норма – 50-200%), фибриноген – 3,4 г/л (норма – 2,0-4,0 г/л), фактор ХIIа зависимый фибринолиз – 80 мин (норма – 5-12 мин), фактор XII – 0% (норма – 70-120%), фактор Виллебранда – 52% (норма – 50-150%). Заключение: при коагулогическом обследовании выявлены отсутствие ФXII, удлинение АЧТВ и замедление фибринолиза. Был установлен диагноз болезни Хагемана (дефицит ФXII). Кроме того, при генетическом исследовании было обнаружено, что пациентка является носителем мутации гена ингибитора активации плазминогена PAI-I (4G/5G), что также приводит к значительному подавлению фибринолиза.
В настоящее время пациентка М. находится под постоянным диспансерным наблюдением врача-гематолога по месту жительства. С учетом имеющейся патологии системы гемостаза пациентке противопоказано назначение ингибиторов фибринолиза (транексамовая кислота), а при наличии показаний к проведению оперативного вмешательства необходимо предварительное проведение трансфузионной подготовки (за 30 мин до операции в/в переливание 1000 мл одногруппной свежезамороженной плазмы на фоне приема Н1-гистаминоблокаторов).
В заключение хотелось бы отметить, что представленное клиническое наблюдение демонстрирует выявление редкого наследственного дефекта системы гемостаза – дефицита фактора XII свертывания крови у пациентки 58 лет. Особенностью данного случая является то, что заболевание длительное время протекало бессимптомно, усугубляя течение кардиоваскулярной патологии, по поводу которой пациентка неоднократно проходила обследование и лечение. Случайное выявление удлинения АЧТВ перед плановым эндоваскулярным вмешательством послужило поводом для проведения дальнейшего диагностического поиска, результатом которого и стало обнаружение болезни Хагемана. Особый интерес представляет тот факт, что у пациентки М. было обнаружено полное отсутствие фактора XII свертывания крови, тогда как в опубликованных ранее наблюдениях описан только дефицит данного фактора [7-9].
Источник