
Хроническое продуктивное гранулематозное воспаление

Хроническая гранулематозная болезнь – это редкая форма первичного иммунодефицита, при которой нарушается бактерицидная функция фагоцитов. Является наследственным заболеванием, в основном передается по Х-сцепленному рецессивному типу. Патология проявляется рецидивирующими инфекциями легких, ЖКТ, кожи и других органов. Для диагностики используется проточная цитометрия, генетическое тестирование, микробиологические исследования. Лечение включает антибиотикотерапию, противогрибковые препараты, иммуномодуляторы, в отдельных случаях рекомендована трансплантация костного мозга.
Общие сведения
Заболевание известно с 1954 г. благодаря ученому C.A. Джейнвею и его коллегам, а генетические причины патологии начали изучаться только в 1980-х гг. Хроническая гранулематозная болезнь (ХГБ) встречается редко, в США ее регистрируют у 1 ребенка на 200-250 тысяч новорожденных. Данные по распространенности в России отсутствуют. Редкость проблемы, недостаточная осведомленность педиатров и терапевтов, сложности в подборе адекватной поддерживающей терапии – все это обуславливает большую актуальность этой хронической наследственной болезни в медицине.

Хроническая гранулематозная болезнь
Причины
Болезнь вызвана генетическими мутациями, которые передаются по наследству. Самым частым вариантом является Х-сцепленная мутация в гене gp91-фокс (60-70%). Болеют мужчины, а женщины являются носителями дефектного гена. Оставшиеся случаи приходятся на аутосомно-рецессивный тип наследования, связанный с генными мутациями p22-фокс, p47-фокс, p67-фокс. Этот подтипом заболевания могут страдать больные обоего пола.
Патогенез
В основе хронической гранулематозной болезни лежит дефицит НАДФН-оксидазного комплекса, который образуется при участии цитозольных и мембраносвязывающих белковых единиц. Вследствие мутации генов образование этих протеинов нарушается, в организме пациента не синтезируются ферменты, способные превращать молекулярный кислород в активные формы – супероксидный анион, перекись водорода, синглетный кислород.
Эти окислители в норме вырабатываются гранулоцитами (фагоцитами), составляют основу кислородозависимого этапа фагоцитоза. Он необходим для переваривания отдельных видов бактерий и грибков, продуцирующих фермент каталазу (например, Klebsiella sp, Candida albicans, Aspergillus niger). Остальные звенья клеточного иммунитета не нарушены, поэтому вирусные и паразитарные инфекции встречаются не чаще, чем в среднем в популяции.
Симптомы
Дети с ХГБ рождаются здоровыми, клинические признаки заболевания у них появляются в первые 2 года жизни. В атипичных случаях симптомы Х-сцепленного варианта хронической патологии манифестируют к 5 годам, а при аутосомно-рецессивном наследовании – к 9 годам. Основным признаком болезни являются инфекции, вызванные каталазоположительными Грам+ (Staphylococcus aureus) или Грам- бактериями (E. coli, Serratia liquefaciens, Klebsiella sp., P. aeruginosa, Proteus sp.).
Первичный иммунодефицит в 70-80% случаев манифестирует с рецидивирующих бактериальных инфекций дыхательных путей: тонзиллитов, бронхитов, пневмоний. Инфекционные заболевания отличаются тяжелым течением, умеренным ответом на стандартную антибиотикотерапию, частым переходом в нагноительные процессы. При этом образуются буллы, абсцессы, гангрена легкого.
В дальнейшем у 75-100% пациентов развиваются хронические гнойные лимфадениты, которые проявляются покраснением, припухлостью, болезненностью лимфоузлов. При поражении подмышечных или паховых узлов беспокоит резкая боль при движениях конечностями. В 24-41% случаев формируются гнойные воспаления печени, у 20-30% больных наблюдаются рецидивирующие желудочно-кишечные инфекции, бактериальные и грибковые поражения костей.
Характерным признаком гранулематозной болезни является генерализованный «бецежит», возникающий как осложнение после проведения первой прививки БЦЖ, проявляющийся спустя 1-2 месяца после иммунизации. Патология связана с недостаточностью клеточного иммунитета при введении ослабленных микобактерий Коха. Также после вакцинации есть вероятность начала остеита – формы костного туберкулеза.
Осложнения
Опасное последствие хронической гранулематозной болезни – аспергиллез легких, который провоцирует до 25% случаев смерти среди пациентов. Абсцессы печени чреваты развитием тяжелого гепатолиенального синдрома. Поражение костной ткани грибковым процессом, особенно в области позвоночника и ребер, сопряжено с массивной диссеминацией возбудителя, отличается крайне неблагоприятным прогнозом.
Типичным осложнением иммунодефицита у страдающих гранулематозной болезнью являются неконтролируемые микробные инфекции, которые распространяются по организму с возникновением сепсиса, септицемии, септического шока. При критическом снижении иммунитета, отсутствии своевременной помощи эти заболевания заканчиваются смертью больного. Также при гранулематозной болезни у детей отмечаются задержка роста, отставание в физическом развитии.
Диагностика
При первичной диагностике устанавливаются общие признаки иммунодефицита, выясняется семейный анамнез, степень риска генетической патологии. При физикальном осмотре патогномоничные проявления хронической гранулематозной болезни не определяются. Для постановки диагноза требуется комплексное лабораторно-инструментальное обследование:
- Цитометрия респираторных смывов. Основной способ диагностики, с помощью которого оценивается уровень продукции активных радикалов кислорода. Он подходит для 100% подтверждения хронической генетической патологии, а также для выявления бессимптомных женщин-носительниц гена при Х-сцепленном варианте наследования.
- Генетическое исследование. Рекомендуется по показаниям, чаще в научных целях при подборе экспериментальных методов лечения. Анализ производится методом секвенирования генома, FISH-гибридизации. На ХГБ указывают мутации в генах CYBB, CYBA, NCF1, NCF2 и NCF4.
- Микробиологическая диагностика. Чтобы выявить каталаза-продуцирующие микроорганизмы, проводится бактериоскопия, бакпосев на питательные среды. Для выбора рациональной антибиотикотерапии выполняется тест на антибиотикочувствительность.
- Инструментальная визуализация. Чтобы обнаружить гнойные поражения органов, назначается УЗИ плевральной и брюшной полостей, лимфатических узлов. Также используются рентгенологические исследования (рентгенография, КТ), эндоскопические методы (бронхоскопия, ЭФГДС, колоноскопия).
Лечение хронической гранулематозной болезни
Консервативная терапия
При острых эпизодах микробных инфекций, возникших у пациентов с ХГБ, назначаются стандартные антибиотики из класса макролидов, цефалоспоринов, фторхинолонов. Для терапии грибкового поражения наиболее эффективен амфотерицин В, который принимается длительными курсами. Поддерживающее лечение, которые направлено на снижение риска инфекционных осложнений, включает следующие лекарства:
- Бактерицидные препараты. Для предупреждения бактериальных заболеваний рекомендован длительный прием сульфаниламидов, макролидов.
- Антимикотики. Применяются, чтобы снизить риск аспергиллеза – наиболее опасной грибковой инфекции при хронической гранулематозной болезни.
- Гамма-интерферон. Иммуномодулятор используется, чтобы повысить устойчивость организма к инфекционным агентам, усилить продукцию супероксидных радикалов нейтрофилами.
Экспериментальное лечение
Наиболее перспективным методом терапии больных хронической гранулематозной болезнью считается трансплантация гемопоэтических стволовых клеток, чтобы полностью восстановить нормальную иммунную защиту. Однако пересадка сопряжена с высоким риском осложнений, поэтому проводится ограничено в рамках исследовательских программ. На стадии разработки находятся различные методы генной терапии.
Прогноз и профилактика
Несмотря на применяемые методы лечения, нередко развиваются системные грибковые и бактериальные инфекции с летальным исходом. Прогноз относительно неблагоприятный, наблюдается ухудшение качества и снижение продолжительности жизни больных. Профилактика осложнений предполагает обеспечение постоянной поддерживающей терапии, проведение вакцинации в полном объеме (за исключением БЦЖ), максимальное ограждение пациента от инфекций.
Источник
В основе гранулематозного воспаления лежат иммунные нарушения – главным образом по типу гиперчувствительности замедленного типа, аллергические и цитотоксические реакции. По мнению А.А. Ярилина (1999), развитие гранулемы, как правило, служит показателем неэффективности иммунной защиты. Появление гранулем в ходе воспалительного процесса нередко связано с несостоятельностью мононуклеарных фагоцитов, которые не могут переварить возбудителя, а также с персистированием последнего в тканях.
В связи с особенностями реакции организма на тот или иной агент гранулематозное воспаление называют также специфическим. Оно характеризуется определенным возбудителем, сменой и полиморфизмом тканевых реакций в соответствии с состоянием иммунной системы организма, хроническим волнообразным течением, преобладанием продуктивной реакции гранулематозного характера и развитием коагуляционного некроза в очагах воспаления. К инфекционным заболеваниям, характеризующимся специфичностью реакции, относят туберкулез, сифилис, лепру, склерому. Воспалительный процесс при этих заболеваниях имеет, как обычно, все компоненты: альтерацию, экссудацию и пролиферацию, но, кроме того, ряд определенных морфологических признаков в виде гранулемы – довольно четко отграниченного скопления гистиоцитов или эпителиоидных клеток в дерме на фоне хронической воспалительной инфильтрации, нередко с примесью гигантских многоядерных клеток.
Эпителиоидные клетки представляют собой разновидность макрофагов, содержат гранулярную эндоплазматическую сеть, синтезируют РНК, но к фагоцитозу мало способны, однако обнаруживают способность к пиноцитозу малых частиц. Эти клетки имеют неровную поверхность из-за большого количества микроворсинок, плотно соприкасающихся с микроворсинками соседних клеток, в результате чего в гранулеме они тесно прилежат друг к другу. Считается, что гигантские клетки образуются из нескольких эпителиоидных клеток благодаря слиянию их цитоплазмы.
Классификация гранулематозного воспаления чрезвычайно трудна. Как правило, при этом основываются на патогенетических, иммунологических и морфологических критериях. W.L. Epstein (1983) делит все гранулемы кожи, в зависимости от этиопатогенетического фактора, на следующие типы: гранулему инородного тела, инфекционные, иммунные, связанные с первичным повреждением ткани и не связанные с тканевыми повреждениями. О. Reyes-Flores (1986) классифицирует гранулематозное воспаление в зависимости от иммунного статуса организма. Он различает иммунекомпетентное гранулематозное воспаление, гранулематозное воспаление с неустойчивым иммунитетом и иммунодефицитное.
А.И. Струков и О.Я. Кауфман (1989) разделили все гранулемы на 3 группы: по этиологии (инфекционные, неинфекционные, медикаментозные, пылевые, гранулемы вокруг инородных тел, неустановленной этиологии); гистологии (гранулемы из зрелых макрофагов, с/без эпителиоидных, или гигантских, многоядерных клеток, с некрозом, фиброзными изменениями и др.) и патогенезу (иммунные гиперчувствительные гранулемы, неиммунные гранулемы и др.).
B.C. Hirsh и W.C. Johnson (1984) предложили морфологическую классификацию, учитывающую выраженность тканевой реакции и преобладание в этом процессе того или иного типа клеток, наличие нагноения, некротических изменений и инородных тел или инфекционных возбудителей. Авторы различают пять типов гранулем: туберкулоидную (эпителиоидно-клеточную), саркоидную (гистиоцитарную), типа инородных тел, некробиотическую (палисадообразную) и смешанную.
Туберкулоидные (эпителиоидно-клеточные гранулемы) встречаются главным образом при хронических инфекциях (туберкулез, поздний вторичный сифилис, актиномикоз, лейшманиоз, риносклерома и др.). Они образованы эпителиоидными и гигантскими многоядерными клетками, среди последних преобладают клетки Пирогова-Лангханса, но встречаются и клетки инородных тел. Для этого типа гранулем характерно наличие широкой зоны инфильтрации лимфоцитарными элементами вокруг скоплений эпителиоидных клеток.
Саркоидная (гистиоцитарная) гранулема представляет собой тканевую реакцию, характеризующуюся преобладанием в инфильтрате гистиоцитов и многоядерных гигантских клеток. В типичных случаях отдельные гранулемы не склонны к слиянию между собой и окружены венчиком из очень небольшого количества лимфоцитов и фибробластов, которые в самих гранулемах не определяются. Гранулемы этого типа развиваются при саркоидозе, внедрении циркония, при татуировке.
Некробиотические (палисадообразные) гранулемы встречаются при кольцевидной гранулеме, липоидном некробиозе, ревматических узелках, болезни кошачьих царапин и венерическом лимфогранулеме. Некробиотические гранулемы могут быть различного генеза, некоторые из них сопровождаются глубокими изменениями сосудов, чаще первичного характера (гранулематоз Вегенера). Гранулема инородных тел отражает реакцию кожи на инородное тело (экзогенное или эндогенное), характеризуется скоплениями вокруг него макрофагов и гигантских клеток инородных тел. В смешанных гранулемах, как следует из названия, сочетаются признаки разных типов гранулем.
Гистогенез гранулематозного воспаления подробно описан D.O. Adams. Экспериментально этот автор показал, что развитие гранулемы зависит от характера вызывающего агента и состояния организма. В начальных фазах процесса появляется массивный инфильтрат из юных мононуклеарных, фагоцитов, гистологически напоминающий картину хронического несиецифического воспаления. Через несколько дней этот инфильтрат превращается в зрелую гранулему, причем агрегаты зрелых макрофагов располагаются компактно, они превращаются в эпителиоидные, а затем в гигантские клетки. Этот процесс сопровождается ультраструктурными и гистохимическими изменениями мононуклеарных фагоцитов. Так, юные мононуклеарные фагоциты представляют собой относительно мелкие клетки, имеют плотные гетерохроматиновые ядра и скудную цитоплазму, в которой содержатся немногочисленные органеллы: митохондрии, комплекс Гольджи, зернистая и гладкая эндоплазматическая сеть и лизосомы. Эпителиоидные клетки крупнее, имеют эксцентрически расположенное эухроматиновое ядро и обильную цитоплазму, содержащую, как правило, большое количество органелл.
При гистохимическом исследовании в мононуклеарных фагоцитах в начале их развития выявляются пероксидазоположительные гранулы, напоминающие таковые в моноцитах, в этпелиоидных клетках отмечаются прогрессирующее растворение первичных пероксидазоположительных гранул и увеличение количества пероксисом. При прогрессировании процесса в них появляются лизосомальные ферменты, такие как бета-галактозидаза. Изменения ядер клеток гранулемы от маленьких гетерохроматиновых до крупных эухроматиновых обычно сопровождаются синтезом РНК и ДНК.
Кроме описанных выше элементов гранулемы, в ней встречаются в различном количестве нейтрофильные и эозинофильные гранулоциты, плазмоциты, Т- и В-лимфоциты. В гранулемах очень часто наблюдается некроз, особенно в случаях высокой токсичности aгентов, вызвавших гранулематозное воспаление, таких как стрептококки, кремний, микобактерии туберкулеза, гистоплазмы. Патогенез некроза в гранулемах точно неизвестен, однако имеются указания на влияние таких факторов, как кислые гидролазы, нейтральные протеазы и различные медиаторы. Кроме того, придают значение лимфокинам, влиянию эластазы и коллагеназы, а также спазмам сосудов. Некроз может быть фибриноидным, казеозным, иногда сопровождается размягчением или гнойным расплавлением (абсцедировоние). Инородный материал или возбудитель в гранулемах. подвергаются деградации, однако они могут вызвать иммунный ответ. Если вредные вещества полностью инактивируются, то гранулема регрессирует с образованием поверхностного рубца.
Если же этого не происходит, то указанные вещества могут находиться внутри макрофагов и отделяются от окружающих тканей фиброзной капсулой или секвестрируются.
Формирование гранулематозного воспаления контролируется Т-лимфоцитами, которые распознают антиген, превращаются в бластные клетки, способные информировать другие клетки и лимфоидные органы, участвуют в процессе пролиферации вследствие продукции биологически активных веществ (интерлейкин-2, лимфокины), называемых макрофагально-активными хемотаксическими факторами.
[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11]
Источник
Хроническая гранулематозная болезнь (ХГБ) – достаточно неплохо изученное на сегодняшний день наследственное заболевание, связанное с недостаточностью фагоцитарной системы, следствием которой является иммунодефицит. Впервые ХГБ была описана в 1959 году у детей, для которых болезнь оказалась смертельной [1]. Болезнь не поддавалась лечению; на аутопсии выявляли генерализованное гранулематозное воспаление, отличающееся по своей специфике от известных на тот момент инфекционных и неинфекционных заболеваний.
Сегодня нам известно намного больше. Заболевание в большинстве случаев более не является смертельным, выявляется в основном у детей (врожденный иммунодефицит проявляется достаточно быстро), неплохо купируется. Однако до сих пор ХГБ относится к заболеваниям неизлечимым.
Этиология и патогенез
Сущность хронической гранулематозной болезни – в недостаточности фермента НАДФ-оксидазы, который представляет собой комплекс каталитических протеинов. Этот фермент, находясь в фагоцитирующих клетках, обеспечивает перенос электрона с НАДФ на молекулярный кислород: происходит т. н. «респираторный взрыв» (см. Рис. 1). Строго говоря, данный фермент есть не только у фагоцитов: единственным специфичным для них белком является трансмембранный gp91, остальные же компоненты встречаются в самых разных клетках [2].
Именно этот респираторный взрыв и позволяет завершить фагоцитоз и лизировать микроб, в противном же случае будет наблюдаться феномен эндоцитобиоза – он же «незавершенный фагоцитоз» – когда микроб просто живет внутри клетки. Фагоцит вполне себе поглотит бактерию, а убить не сможет. Таков механизм развития многих тяжелых инфекций (например, туберкулеза) [3]; эта же патология лежит в основе и других первичных иммунодефицитов [4].
Обратимся к рисунку 1. Белки gp91 и p22 объединяют в цитохром b558 – мембраносвязанную часть фермента НАДФ-оксидазы, остальные протеины называют цитозольными. При активации фагоцита различными медиаторами цитозольные p47 и p67 фосфорилируются и связываются вместе. Комплекс приобретает сродство к белкам p47 и rac2 – таким образом, присоединяясь к ним, данные протеины вызывают конформационные изменения в мембранном цитохроме b558, – и комплекс приобретает оксидазную активность [2, 5]. НАДФ-оксидаза переносит электрон от своего кофермента НАДФ на кислород с формированием активных форм кислорода (АФК) – O2- и H2O2. Вот здесь и начинается самое интересное.
Классически считается, что фагоцит убивает микробы, образуя фаголизосому с бактерией или грибом, воздействуя на них большими дозами высокотоксичных АФК; однако в последнее время приобретает актуальность иная точка зрения. В 2002 году в Nature была опубликована статья, авторы которой пересмотрели всю парадигму деактивации поглощенных микробов.
Ученые обнаружили, что если в фагоцитах мышей определяется нормальный уровень активных форм кислорода, но имеется недостаточность лизосомальных ферментов – животные будут беззащитны против стафилококковых и кандидозных инфекций. То есть, несмотря на наличие нормального респираторного взрыва, иммунодефицит все равно присутствует. Стало быть, эффекторами в инактивации микроба являются не сами АФК [6].
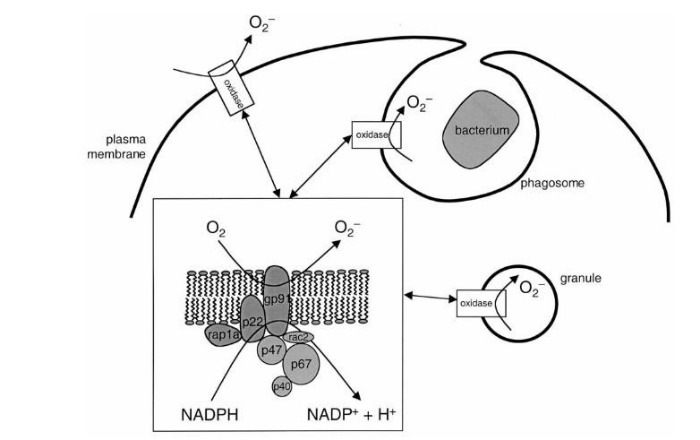 Рисунок 1. Механизм формирования NADPH-оксидазы [5].
Рисунок 1. Механизм формирования NADPH-оксидазы [5].
Согласно новой модели, АФК являются медиаторами в уничтожении микроба, а непосредственные «убийцы» – ферменты лизосом, обладающие протеолитической активностью. Как известно, при захвате фагоцитом микроба формируется вакуоль – (фагосома), с которой впоследствии сливается лизосома макрофага [7]. На мембране этой вакуоли и функционирует НАДФ-оксидаза, постоянно увеличивая концентрацию АФК. Однако как уже было сказано, данный процесс – не конечный эффекторный механизм.
Предположительно, супероксид-анион (O2-) вызывает приток ионов калия. K+, в свою очередь, приводит pH к оптимальным для функционирования протеолитических ферментов показателей. В роли калиевых каналов, возможно, может выступать как обычный протонный канал, так и сам комплекс НАДФ-оксидазы (который в данном случае будет представлен в качестве белка-переносчика) [6, 8].
Наследование и генетика
Хроническая гранулематозная болезнь имеет наследственную природу. Приблизительно данной патологией страдает 1 на 250 000 [9], что делает заболевание достаточно редким и потому трудным в диагностическом отношении. Заболевание вызывает мутация любого из четырех генов, кодирующих субъединицы ключевого фермента фагоцитоза НАДФ-оксидазы. Более двух третей случаев связаны с X-сцепленным наследованием (дефект гена CYBB, кодирующего белок p-91); остальные случаи связаны с аутосомно-рецессивным наследованием генов CYBA, NCF-1 и NCF-2, кодирующих белки p22, p47 и p67 соответственно. Исходя из этого болезнь обозначают как ХГБ X91, A22, A47 и A67 (в зависимости от типа наследования и локуса гена) [10].
Примечательно, что в литературе нет (или крайне мало) доказанных случаев наследственного дефекта других субъединиц. Однако в последнее время обнаруживаются все новые мутации генов, следствием которых становится ХГБ: например, в 2009 году выделили еще один подвид ХГБ, связанный с аутосомно-рецессивной мутацией гена p-40 [11], имеются также сведения о единственном пациенте с недостаточностью белка Rac2 – [10]. Вполне вероятно, что могут существовать и другие генетические патологии, вызывающие данное заболевание.
Диагностика
Диагностика ХГБ основана на выявлении клинических признаков, кроме того, важно выявить наличие или отсутствие респираторного взрыва. Последнее можно осуществить несколькими гистологическими и иммунологическими методами, например, окрашивание нитросиним тетразолием (НТЗ) позволяет определить, вырабатывают ли клетки АФК – НТЗ будет утилизироваться активными формами кислорода, в результате чего из бледно-желтого тетразолия образуется голубой формазан [12].
Среди наиболее ярких клинических симптомов можно выделить: пиодермию, пневмонию, воспалительные процессы желудочно-кишечного тракта, лимфаденит, абсцесс печени и остеомиелит [13] на фоне рецидивирующих бактериальных и грибных инфекций.
В крови выявляется гипергаммаглобулинемия и анемия. В местах дренажей – хроническое воспаление с образованием гранулем. Также гранулемы могут формироваться в различных тканях и органах: например, в желудке гранулематозное воспаление способно привести к обструкции желудочного канала, в урогенитальном тракте – к циститу. Также следует упомянуть, что почти 20 % больных ХГБ страдает от гранулематозного колита, который легко перепутать с болезнью Крона [14].
Основную же опасность для жизни пациента представляют различные инфекции. Примечательно, что поражают организм достаточно небольшой спектр бактерий и грибов: Staphylococcus, Burkholderia, Serratia, Nocardia, и некоторые грибы рода Aspergillus, реже – Salmonella и Mycobacterium tuberculosis [15, 16]. Несколько реже встречается инфицирование Chromobacterium violaceum (бацилла семейства Neisseriaceae, вызывающая тяжелые инфекционные осложнения в виде кожных фурункулов, абсцессов внутренних органов и септического поражения) [17]. Все они требуют специфического лечения. Кроме того, поражение органов также может быть стереотипным (см. Рис.2).
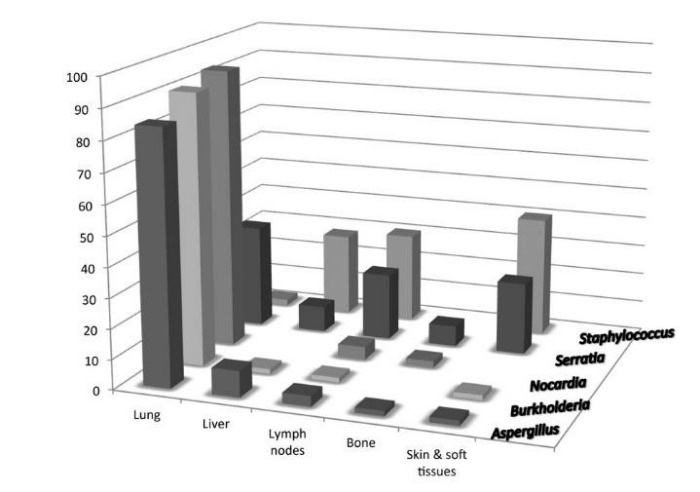 Рисунок 2. Сравнительная характеристика частоты органных поражений при некоторых инфекциях у пациентов с ХГБ [15].
Рисунок 2. Сравнительная характеристика частоты органных поражений при некоторых инфекциях у пациентов с ХГБ [15].
Также следует упомянуть о недавно открытой бактерии, выявленной у пациентов с ХГБ, которую исследователи предложили назвать Granulobacter bethesdensis. Это грамотрицательная палочка, которая на данный момент не может быть отнесена ни к одной из существующих таксономических групп; бактерия поражает лимфоузлы, кроме того, резистентна к антибиотикам in vitro и, скорее всего, – in vivo [18, 19]. Предполагается, что это – лишь первая из подобных бактерий, возникшая в эру антибиотиков. На данный момент Granulobacter bethesdensis не слишком распространена, однако имеет к этому весьма опасную тенденцию.
Лечение
В настоящее время активно разрабатываются методики, которые смогут не просто позволить больным с ХГБ жить полноценно, но и в перспективе совершенно избавить их от бремени заболевания. Поскольку болезнь наследственная, крайне трудно придумать что-то существенное, однако такие попытки предпринимаются, и некоторые из них обнадеживают.
Одна из них – лечение хронической гранулематозной болезни с помощью генной инженерии [20]. В 2006 году в Nature Medicine была опубликована статья, авторы которой сообщили об успешной коррекции генома двух пациентов с X-сцепленной формой ХГБ. После лечения у пациентов определяли активность нейтрофилов с помощью позитронно-эмиссионной томографии, а также других инструментальных методов. Исследование показало, что в обоих случаях фагоциты после проведенного лечения смогли оказать сопротивление инфекции. На данный момент это – одна из самых многообещающих методик.
Еще одним способом терапии является пересадка гемопоэтических клеток [21]. Двадцати семи пациентам после миелоаблативного режима кондиционирования (подготовка пациента к трансплантации с помощью лучевой или цитостатической терапии – прим. автора) пересадили гемопоэтические стволовые клетки от наиболее подходящих доноров (по белкам HLA – главного комплекса гистосовместимости). Двадцать три пациента вполне успешно перенесли операцию, а дальнейшее наблюдение позволило говорить об излечении этих пациентов от хронической гранулематозной болезни. Однако еще 4 пациента умерли от последующих инфекций (15 %).
В остальном же современная медицина может предложить крайне немного. Это – патогенетическая и симптоматическая терапия с использованием антибиотиков, дренажей и прочего. Излишне говорить, что подобное лечение не способно избавлять пациентов от ХГБ.
Профилактика инфекций.
Крайне важно предотвращение развития инфекционного процесса. Пациенты с ХГБ испытывают невероятные трудности в повседневной жизни, например, некачественная чистка зубов спокойно может окончиться гингивитом, а царапина – тяжелой бактериемией. Потому больным необходимо тщательным образом следить за гигиеной, выполнять профессиональную чистку зубов, обрабатывать все царапины антисептиком. И, разумеется, такие пациенты должны быть привиты по всем правилам [14]. В противном же случае даже достаточно простая инфекция может окончиться летально.
Источники:
- R. A. BRIDGES, H. BERENDES, and R. A. GOOD, “A Fatal Granulomatous Disease of Childhood,” AMA. J. Dis. Child., vol. 97, no. 4, p. 387, 1959.
- S. M. Holland, “Chronic Granulomatous Disease,” Clin. Rev Allerg Immunol, no. June 2009, pp. 3-10, 2010.
- B. D. L. Clemens and M. a Horwitz, “Characterization of the Mycobacterium mberctdosis Phagosome and Evidence that Phagosomal Maturation Is Inhibited,” Culture, vol. 181, no. January, 1995.
- W. Al-Herz et al., “Primary immunodeficiency diseases: an up on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency,” Front. Immunol., vol. 2, no. November, pp. 1-26, 2011.
- C. Dahlgren and A. Karlsson, “Respiratory burst in human neutrophils Claes,” J. Immunol. Methods, vol. 232, pp. 3-14, 1999.
- E. P. Reeves et al., “Killing activity of neutrophils is ted through activation of proteases by K+flux,” Nature, vol. 416, no. 6878, pp. 291-297, 2002.
- A. Aderem and D. M. Underhill, “Mechanisms of Phagocytosis in Macrophages,” Annu. Rev. Immunol., vol. 17, no. 1, pp. 593-623, 1999.
- A. Maturana et al., “Heme Histidine Ligands within gp91phox Modulate Proton Conduction by the Phagocyte НАДФ Oxidase,” J. Biol. Chem., vol. 276, no. 32, pp. 30277-30284, 2001.
- J. M. van den Berg et al., “Chronic granulomatous disease: The European experience,” PLoS One, vol. 4, no. 4, pp. 1-10, 2009.
- P. G. H. Ãy, A. R. Cross, and J. T. C. Ã, “Chronic granulomatous disease,” no. 15, pp. 578-584, 2003.
- J. D. Matute et al., “A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil НАДФ oxidase activity,” Blood, vol. 114, no. 15, pp. 3309-3316, 2009.
- M. D. . Brahm H. Segal, P. D. . Thomas L. Leto, M. D. . John I. Gallin, M. D. . Harry L. Malech, and M. D. Steven M. Holland, “Genetic, biochemical, and clinical features of chronic granulomatous disease.” 2000.
- J. A. Winkelstein et al., “Chronic Granulomatous Disease.” 2000.
- R. A. Seger, “Modern management of chronic granulomatous disease,” Br. J. Haematol., vol. 140, no. 3, pp. 255-266, 2008.
- B. E. Marciano et al., “Common severe infections in chronic granulomatous disease,” Clin. Infect. Dis., vol. 60, no. 8, pp. 1176-1183, 2015.
- N. Bennett, P. J. Maglione, B. L. Wright, and C. Zerbe, “Infectious Complications in Patients With Chronic Granulomatous Disease,” vol. 7, no. June, 2018.
- Z. Meher-Homji, R. P. Mangalore, P. D. R. Johnson, and K. Y. L. Chua, “Chromobacterium violaceum infection in chronic granulomatous disease: a case report and review of the literature,” JMM Case Reports, vol. 4, no. 1, 2017.
- D. E. Greenberg et al., “A novel bacterium associated with lymphadenitis in a patient with chronic granulomatous disease,” PLoS Pathog., vol. 2, no. 4, pp. 260-267, 2006.
- D. E. Greenberg et al., “Recurrent granulibacter bethesdensis infections and chronic granulomatous disease,” Emerg. Infect. Dis., vol. 16, no. 9, pp. 1341-1348, 2010.
- M. G. Ott et al., “Correction of X-ed chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1,” Nat. Med., vol. 12, no. 4, pp. 401-409, 2006.
- R. A. Seger et al., “Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: A survey of the European experience, 1985-2000,” Blood, vol. 100, no. 13, pp. 4344-4350, 2002.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник